
510k认证和fda认证
510k认证和fda认证
对美国的器械出口数量逐年增长,越来越多的企业需要面对美国食品与药品监督管理局FDA的审核。对于高风险的器械,通常需要向美国FDA提交510(k)申请(也叫上市前通知),在获得510(k)批准以后,方可销售到美国。往往510(k)申请是企业需要面对的510(k)申请显 一大难题。相比CE认证,美国FDA的更加灵活、往往企业遇到的困难更大。在深入理解美国FDA相应法规要求的510(k)历年申请情况进行 基础上,对美国FDA的分析和总结是非常有必要的。
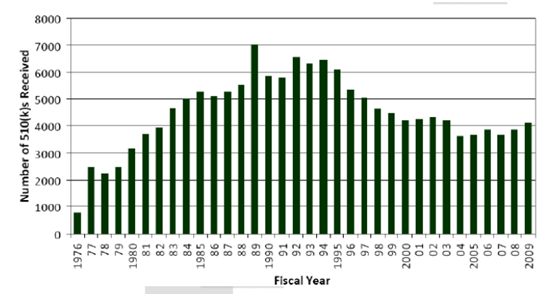
美国FDA从1976年到2009年中每一年收到的510(k)申请数量。从图中可以看出,80年代末和90年代初510(k)的申请达到了高峰(每年6000-7000件申请),之后出现缓慢下降,近几年处于比较平稳的状态(每年接近4000件申请)。我们从这些数字可以看出当时美国FDA关于510(k)的一些政策变化。1989年的510(k)申请有非常显著的增加,这种增加Zui主要是由于FDA对检查类手套从510(k)豁免变成了非豁免要求所造成的。这个法规要求变化以后,美国FDA一时间收到了超过1000件的已经投入市场的检查类手套510(k)申请。510(k)申请数量在90年代下降的原因是FDA豁免了大部分的一类器械以及1997年通过的食品药品管理局现代化法(FDAMA)让几十种二类器械获得了510(k)豁免。为了进一步对美国FDA工作量进行管理以及更加合理的分配资源,美国对于那些不需要符合上市前通知要求即可保证安全性和有效性的一类器械产品进行了510(k)豁免。在这个背景下,在1976年通过的器材修正案(Medical Device Amendments)和1997年通过的食品药品管理局现代化法(FDAMA)期间,美国FDA 一共对574个一类器械产品豁免了。
510(k) FDAMA的通过使绝大部分一类器械产品获得了510(k)豁免,除了那些预期用途是为了防止危害健康方面及其重要或者具有造成生病或受伤等重大潜在风险的产品(“保留”条件)。因此,一类器械产品中只有那些符合保留条件的产品需要遵守510(k)上市前通知的要求。
510k认证和fda认证
同时FDAMA也授权FDA直接对某些二类器械产品进行510(k)豁免。1998年1月21日,FDA公布了二类器械510k(k)豁免名单,参见法规63 FR 3142。随着2002年器械使用费现代化法(MDUFMA)的通过,510(k)申请数量再一次下降。Zui近几年又有少量回升的趋势。 我们预计将来会有更多的二类器械获得510(k) 豁免。
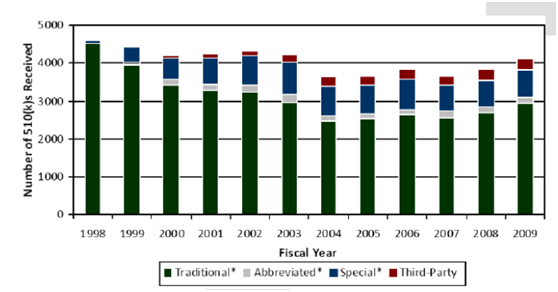
美国FDA从1998年到2009年中每一年收到的不同类别的510(k)申请数量。从图中可以看出,Traditional类型的510(k)数量有所减少,Abbreviated类型的510(k)数量基本维持,Special类型的510(k)数量有所增加,第三方审核的510(k)数量有微小上升。 我们从这些数字可以看出510(k)申请类别仍然以Traditional和Special的方式为主,Abbreviated以及第三方审核仍然占据很少的份额。
510k认证和fda认证
- 510k认证内容 2024-10-20
- 510k注册认证第三方 2024-10-20
- 510k认证时间要多久 2024-10-20
- 亚马逊510k认证 2024-10-20
- 510k认证机构 2024-10-20
- 美国510k认证 2024-10-20
- 导尿管510k认证办理介绍 2024-10-20
- MSDS鉴定书编号 2024-10-20
- msds是运输条件鉴定书 2024-10-20
- 有MSDS没有海运鉴定书 2024-10-20
- msds和化工品鉴定书 2024-10-20
- 货运鉴定书MSDS 2024-10-20
- msds和货物运输鉴定书 2024-10-20
- msds鉴定书怎么做 2024-10-20
- 化工品鉴定书是msds吗 2024-10-20
